Metabolic requirements of pancreatic beta cell proliferation
Contact PI: Matthew Wortham, PhD, University of Colorado Anschutz Medical Campus (R03 DK135459)
Start Date: May 1, 2023
*Moved from CTAR to CBDS on November 1, 2023
NIH HIRN Gateway Investigator Award Recipient
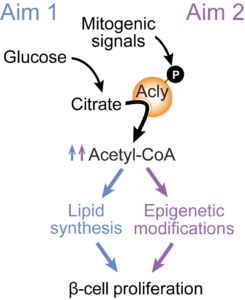
Abstract
Type 1 diabetes (T1D) results from autoimmune destruction of pancreatic β-cells. Potential therapeutic approaches to curtail or reverse the loss of β-cells include immune modulation, β-cell regeneration, or combinations thereof. There has been substantial preclinical development of therapies that stimulate human β- cell replication. However, absolute proliferation rates induced by such drugs remain relatively low, and it is unclear whether current approaches will sufficiently expand β-cell numbers to control blood glucose without exogenous insulin. Here, we build upon emerging evidence that cell metabolism supports proliferation to identify and exploit metabolic bottlenecks that limit therapeutic β-cell expansion. Proliferating cells are known to activate metabolic programs that produce cellular building blocks and generate signals that support cell division. In preliminary data, we found that the metabolic enzyme ATP-citrate lyase (Acly) is required for expansion of transformed β-cells. Other groups have shown that pancreatic deletion of Acly results in smaller islets, consistent with a role for this enzyme in β-cell proliferation. Acly acts upon citrate to produce acetyl-CoA, which is a substrate for lipid synthesis and histone acetylation. In this way, Acly couples mitochondrial metabolism to production of cellular building blocks and signaling via the epigenome. In β-cells, Acly deletion results in downregulation of lipid synthesis genes concomitant with reduced histone acetylation of their associated regulatory elements, suggesting Acly regulates lipid synthesis through dual metabolic and epigenetic mechanisms. Together, these observations build the model that Acly produces both anabolic and signaling metabolites that support β-cell proliferation. We found that Acly is phosphorylated by mitogenic signals, suggesting it could be activated during β-cell proliferation to drive requisite changes in metabolism. Our central hypothesis is that mitogens remodel β-cell lipid metabolism through Acly, and that this effect is necessary for optimal β-cell proliferation. Here, we will test how changes to islet lipid metabolism support β-cell proliferation in primary human islets using genetic or metabolic interventions coupled with targeted metabolomics and genomic assays. In Aim 1, we will monitor the effect of pro-proliferative drugs on islet lipid synthesis and test whether metabolites affected by mitogenic signals limit β-cell proliferation. To assess the requirement for lipid synthesis pathways downstream of Acly, we will determine how ACLY inactivation impacts human β-cell proliferation under the same conditions. In Aim 2, we will assess how the islet epigenome is remodeled in response to β-cell mitogens. Islets will be treated with pro-proliferative drug combinations, then we will perform ChIP-seq for histone modifications previously implicated in governing β-cell proliferation. We will also determine the epigenetic signaling function of Acly by testing its role in nutrient-stimulated histone acetylation in human islets. Understanding the metabolic pathways that support or augment pharmacological β-cell expansion could inform nutritional interventions to optimize β-cell regeneration by therapies currently in preclinical development.