Lactate-mediated Metabolic Reprogramming of Beta Cells in T1D Contributes to their Enhanced Plasticity and Dedifferentiation
Contact PI: Lori Sussel, PhD, University of Colorado Anschutz Medical Campus
Start Date: September 2022
End Date: June 2025
HIRN Catalyst Award Recipient
Abstract
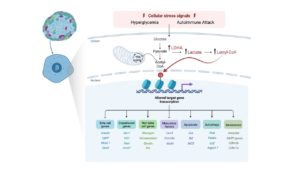
Beta cell failure is associated with both type 1 diabetes (T1D) and type 2 diabetes (T2D). Traditionally, cell death was thought to be the primary mechanism underlying beta cell failure; however in recent years, there is compelling evidence that beta cell loss can also be associated with loss of beta cell identity. This HIRN Catalyst proposal explores a novel molecular mechanism that has the potential to link the changes in metabolic function that occur in the context of diabetes with the gene regulatory programs that disrupt beta cell identity. The study is based on a recent discovery in cancer cells that identified a novel histone posttranslational modification – lysine lactylation – that is induced by increased cellular lactate levels. Interestingly, adult islet beta cells, by virtue of their unique metabolic status, normally express low levels of LDHA and have limited lactate production. In humans and mouse models of diabetes it has been demonstrated that the disallowed gene Ldha becomes highly upregulated in beta cells to contribute to metabolic dysfunction. We hypothesize that the ensuing increase in lactate levels in a diseased beta cell induces histone lactylation to alter the global epigenetic landscape and transcriptional program to trigger the loss of beta cell identity. Understanding how histone lactylation in the stressed beta cell alters identity and function could reveal novel therapeutic strategies to restore beta cell identity and function in T1D.