Targeting ChREBPbeta to Protect Beta-Cells from Metabolic and Inflammatory Stress in T1D
Contact PI: Liora Katz, PhD, Icahn School of Medicine at Mount Sinai (R03 DK147337)
Start Date: May 1, 2026
NIH HIRN Gateway Investigator Award Recipient
Abstract
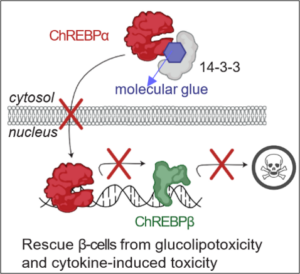
Type 1 Diabetes (T1D) results from progressive destruction of pancreatic β-cells driven by autoimmune attack and intrinsic stress. While much attention has focused on immune-mediated mechanisms, the contribution of β- cell–intrinsic stress pathways to disease progression remains incompletely understood. Emerging evidence implicates ChREBPβ (Carbohydrate-Responsive Element-Binding Protein β), a stress-inducible transcription factor, as a central integrator of metabolic and inflammatory signals that impair β-cell identity and survival. Although ChREBP has been studied in the context of glucose metabolism and Type 2 Diabetes, its pathological role in T1D remains unexplored. Our preliminary data demonstrate that ChREBP and its target genes are upregulated in β-cells from autoantibody-positive (AAb+) and T1D donors, as well as in NOD mice, suggesting early activation in disease pathogenesis. ChREBPβ overexpression induces apoptosis, oxidative stress, and β-cell dedifferentiation. We also developed a novel small molecule, Compound 43, which functions as a “molecular glue” that stabilizes the ChREBPα–14-3-3 interaction, thereby suppressing ChREBPβ expression and protecting β-cells from stress- induced damage, and we demonstrate here that this stabilizer is able to protect human β-cell identity and function under cytokine-induced toxicity. This proposal aims to define the contribution of ChREBPβ to β-cell dysfunction in T1D and evaluate the therapeutic potential of Compound 43 in mitigating this process. In Aim 1, we will characterize the impact of ChREBPβ activation on β-cell stress and survival under inflammatory and ER stress conditions using human islets. We will assess gene expression, apoptosis, UPR activation, and lipid metabolism using transcriptomic and lipidomic profiling. In Aim 2, we will test whether Compound 43 can protect human islets and stem cell– derived β-cells from cytokine- and ER stress–induced dysfunction, using functional, metabolic, and transcriptomic readouts. This study will establish ChREBPβ as a previously unrecognized contributor to early β-cell failure in T1D and provide preclinical validation for a new pharmacologic approach that targets β-cell resilience, rather than immune modulation. These findings will offer a new conceptual and therapeutic framework for preserving β-cell function in the earliest stages of T1D, directly aligning with the mission of the Human Islet Research Network (HIRN) to understand and prevent β-cell failure.