Beta Cell Death in early Type 1 Diabetes caused by Innate Immune Response to Inefficient RNA Editing
Contact PI: Klaus Kaestner, PhD, University of Pennsylvania (U01 DK143477)
Qin Lin, PhD, Investigator, University of Pennsylvania
Yuval Dor, PhD, Consultant, Hebrew University Medical School
Agnes Klochendler PhD, Consultant, Hebrew University Medical School
Start Date: August 1, 2025
*Moved from CBDS to HPAC on May 1, 2026
Abstract
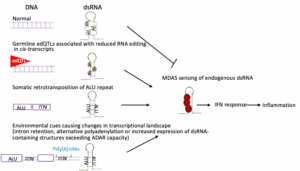
More than 8 million people worldwide are afflicted with type 1 diabetes (T1D), a disease that despite improved insulin treatment still results in a reduction of life expectancy by 12 years. The current model for T1D progression posits that in a subset of individuals with genetic susceptibility, the disease is precipitated by an as yet ill-defined triggering event. The nature of this triggering event remains a crucial open question in T1D. Viral infections have been extensively studied as precipitating events, but no causative viruses have been proven to date. Importantly, an anti-viral interferon response in recent onset T1D patients is well documented. However, this response can be activated not only by viral infection, but also by insufficient adenine to inosine editing of endogenous double stranded RNAs. Recent work from our team has shown that loss of adenine to inosine editing in mouse beta cells or in human islets causes sterile activation of a massive interferon response, and, in mice, beta cell destruction, while ablation of the critical editing enzyme from pancreatic alpha cells has no effect, supporting a unique susceptibility of beta cells to unedited double stranded RNAs. In addition, we demonstrated that genetic variants that reduce adenine to inosine editing in the human pancreas, so called editing quantitative trait loci or edQTLs, are associated with an increased interferon response and elevated risk of T1D. In this project, we are testing the hypothesis that the interferon response that characterizes early-stage T1D does not result from viral infection, but rather from insufficient editing of endogenous double stranded RNAs. We will evaluate three non-exclusive paths to a sterile IFN response in human islets. Firstly, we will test the impact of germline genetic variation on editing of cis transcripts. Secondly, we will build a dsRNA burden model to quantitively predict the amount of insufficiently edited dsRNAs as a trigger of the IFN response. Thirdly, we will determine if disruption of mRNA homeostasis via metabolic stress, ER stress, or abnormal splicing causes accumulation of double stranded RNAs that exceed the beta cell’s capacity for editing, thus resulting in activation of the interferon response. In sum, the proposed studies have the potential to lead to a paradigm shift in our understanding of the earliest events in the pathogenesis of T1D.